LOFTK (Loss-of-Function ToolKit)
![]()
This readme
This readme accompanies the paper “LOFTK: a framework for fully automated calculation of predicted Loss-of-Function variants.” by Alasiri A. et al. bioRxiv 2021.
Background
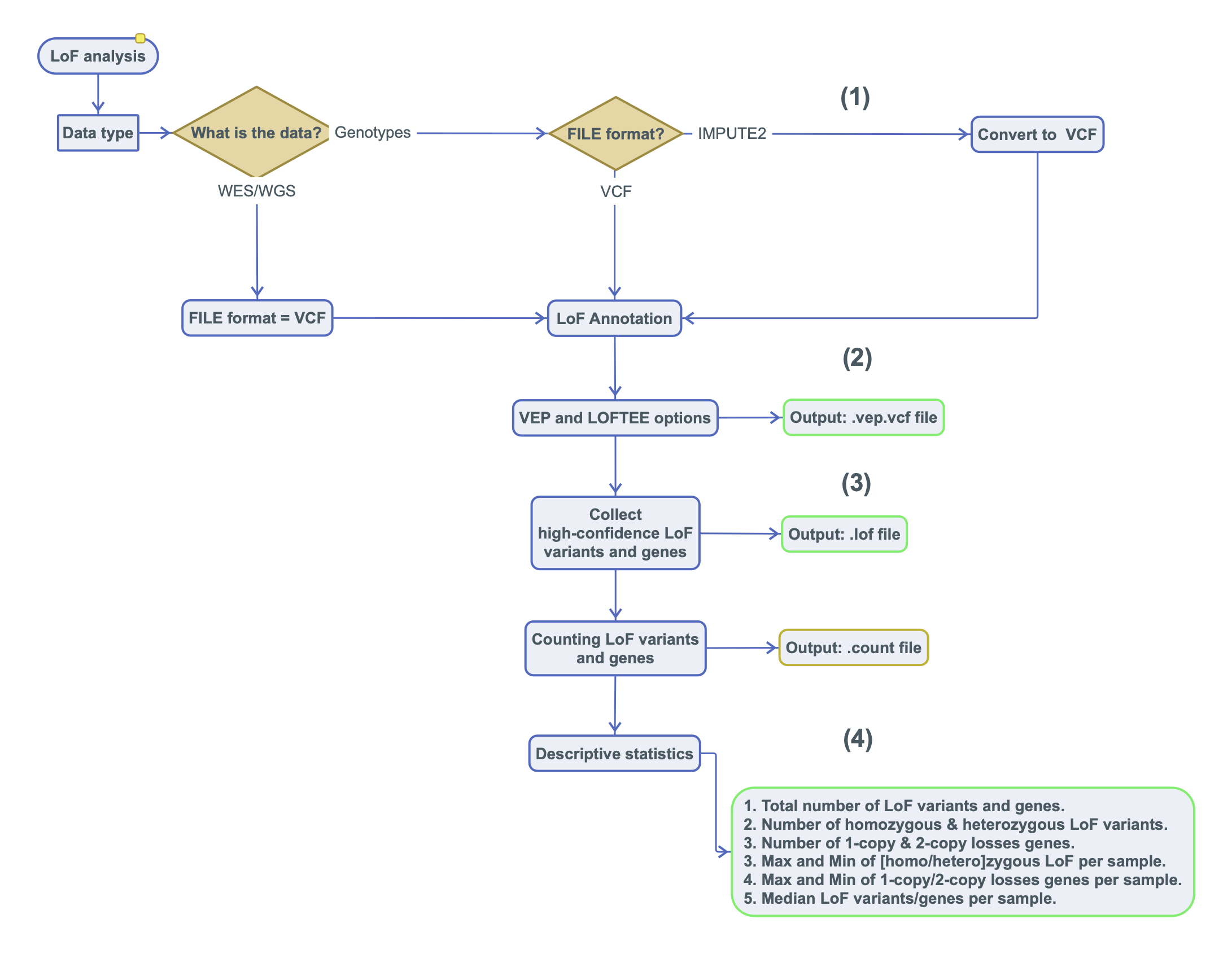
Predicted Loss-of-Function (LoF) variants in human genes are important due to their impact on clinical phenotypes and frequent occurrence in the genomes of healthy individuals. Current approaches predict high-confidence LoF variants without identifying the specific genes or the number of copies they affect. Here we present an open source tool, the Loss-of-Function ToolKit (LoFTK), which allows efficient and automated prediction of LoF variants from both genotyped and sequenced genomes, identifying genes that are inactive in one or two copies, and providing summary statistics for downstream analyses.
LoFTK is a pipeline written in the BASH and Perl languages to identify loss-of function (LoF) variants using VEP and LOFTEE efficiently. It will aid in annotating LoF variants, select high confidence (HC) variants, state the homozygous and heterozygous LoF variants, and calculate statistics.
The Loss-of-Function ToolKit Workflow: finding knockouts using genotyped and sequenced genomes.
Installation and Requirements
Install LoFTK
LoFTK has been developed to work under the environment of two cluster managers; Simple Linux Utility for Resource Management (SLURM) and Sun Grid Engine (SGE). Each cluster manager (SLURM/SGE) has LoFTK verison for installation. Look at Instillation and Requirements in the wiki.
Requirements
All scripts are annotated for debugging purposes - and future reference. The scripts will work within the context of a certain Linux environment - in this case we have tested LoFTK on CentOS7 with a SLURM Grid Engine background.
Perl >= 5.10.1Bash- Ensembl Variant Effect Predictor (VEP)
LOFTEEfor GRCh37- Ancestral sequence
(human_ancestor.fa[.gz|.rz]) - PhyloCSF database
(phylocsf.sql)for conservation filters
- Ancestral sequence
LOFTEEfor GRCh38- GERP scores bigwig
(gerp_bigwig) - Ancestral sequence
(human_ancestor_fa) - PhyloCSF database
(loftee.sql.gz)
- GERP scores bigwig
samtools(must be on path)
Usage
The only script the user should use is the run_loftk.sh script in conjunction with a configuration file LoF.config. It is required to set up the configuration file LoF.config before run any analysis, follow the instruction in the wiki.
You can run LoFTK using the following command:
bash run_loftk.sh $(pwd)/LoF.config
Always Remember
- To set all options in the
LoF.configfile before the run - To use the full path to the configuration file, e.g. use
$(pwd). - You can run LoFTK steps all in one run or separately by setting analysis type in the
LoF.configfile. - VEP and LOFTEE options can be added and modified in one of these configuration files in
./bin/:
Description of files
| File | Description | Usage |
|---|---|---|
| README.md | Description of project | Human editable |
| LICENSE | User permissions | Read only |
| LoF.config | Configuration file | Human editable |
| run_loftk.sh | Main LoFTK script | Read only |
| LoF_annotation.sh | Annotation of LoF variants/genes | Read only |
| allele_to_vcf.sh | Converting IMPUT2 format to VCF | Read only |
| descriptive_stat.sh | Descriptive analysis | Read only |
Post LoFTK
Merge the counts files of multiple cohorts
This scripts allows you to merge the counts files of different cohorts. By default it only includes genes that were present in both files but you can use the union function to include genes that are present in at least 1 cohort. This means that for the other cohorts, the gene LoF counts will be set to 0 for every individual (which is tricky if the gene was not tested), or to a self-specified value
perl merge_gene_lof_counts.pl -i cohortX.counts,cohortY.counts,cohortZ.counts -o merged_cohorts.counts -c
Run the the following to know how to use options:
perl merge_gene_lof_counts.pl --help
Mismatched genes between samples
This script can be used to determine ‘mismatched’ genes between samples; these are genes that are active in one or two copies in one sample and completely inactive (two-copy loss) in the other sample. This feature helps study interactions between human genomes, for instance during pregnancy (maternal vs fetal genome) and after stem cell or solid organ transplantation (donor vs recipient genome).
- You must create a file
pairs_file.txtwith two columns (tab-separated), where both columns have list of individual IDs and each line has paired subjects. - The first column must contain individual IDs for which you want to examine the mismatch of knocked out genes with the 1 or 2 active copies in the other pair.
- Output file contains encodings for individuals (from 1st column in
pairs_file.txt), where1for mismatch0for not mismatch.1: mismatch where sample in the 2nd column has active gene.0: not mismatch where paired samples either having both a knocked out gene or none of them carry LoF gene.
Run the below command:
perl gene_lof_counts_to_dyad_lofs.pl pairs_file.txt input_file.counts output_file.dyads
Inputs
LoFTK permits two common file formats as an input:
-
Variant Call Format (VCF)
You can find VCF specification here. -
IMPUTE2 output format
Four files with the following extensions are needed as an input;.haps.gz,.allele_probs.gz,.infoand.sample
:warning: The input data have to be phased to annotate compound heterozygous LoF variants, which result in LoF genes with two copies losses.
For more details and examples about input files are explained in the wiki.
Outputs
LoFTK will generate four files as an output at the end of the analysis. The LoFTK outputs in the wiki contains more explanation.
- [project_name]_snp.counts: LoF variants and individuals.
- [project_name]_gene.counts: LoF genes and individuals.
- [project_name]_gene.lof.snps: list of LoF variants allele frequencies.
- [project_name]_output.info: report descriptive statistics on LoF variants and genes.
Changes log
Version: v1.0.0</br> Last update: 2021-06-08</br>
- v1.0.0 Initial version.
- v1.0.1
- Add post LoFTK analysis
- Merge and Mismatched genes.
- Add post LoFTK analysis
- v1.0.2
- separate stat description from annotation script
- v1.0.3
- Run each step in LoFTK separately
- Add configuration file to modify VEP and LOFTEE options
Contact
If you have any suggestions for improvement, discover bugs, etc. please create an issues. For all other questions, please refer to the last author:
| Jessica van Setten, PhD | j.vansetten [at] umcutrecht.nl |
CC-BY-SA-4.0 License
| ##### Copyright (c) 2020 University Medical Center Utrecht Creative Commons Attribution-ShareAlike 4.0 International Public License By exercising the Licensed Rights (defined in the [LICENSE](/LoFTK/LICENSE)), you accept and agree to be bound by the terms and conditions of this Creative Commons Attribution-ShareAlike 4.0 International Public License ("Public License"). To the extent this Public License may be interpreted as a contract, you are granted the Licensed Rights in consideration of your acceptance of these terms and conditions, and the Licensor grants you such rights in consideration of benefits the Licensor receives from making the Licensed Material available under these terms and conditions. The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software. THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE. Reference: https://choosealicense.com/licenses/cc-by-sa-4.0/#. |